
Protocols and Starting Points
Here, we provide some favorite protocols and starting points for different types of experiments
This section is like an FAQ for experimental details. Obviously, there are a lot of nuances to every biological question and experiment. However, hopefully this page can give you a set of starting conditions to try a particular type of experiment, and some idea how, and what, to optimize for.
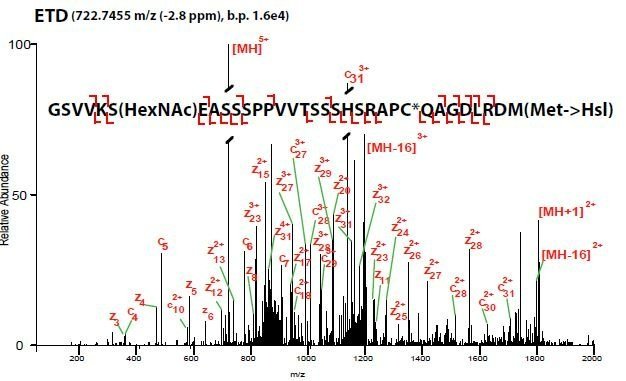
PTM mapping
ETD mass spectrum of an O-GlcNAc modified peptide of SOX2 purified from mESCs and digested in-gel with CNBr.
PTM mapping is isolating your protein of interest (POI) and looking for various PTMs, while estimating their stoichiometry. For these, we like in-gel digestions. They are nearly dummy-proof and often provide very clean results. If you can see a band with a silver stain (yes, their MS compatible) then you should be able to identify appreciably-leveled PTMs. If you have a Coomassie band, you can do other digests than just ol’ reliable trypsin.
This can be done in solution but we find its not quite as sensitive. However, if you think your protein might be modified by Ubiquitin or SUMO, you may want to consider that when resorting to gels.
Protein-protein interactions

Example of Immunoprecipitation followed by western blot. A) Perfect. B) Your protein is still in the flowthrough (FT). Thus, the IP didnt work. C) Couple issues, severity depends on downstream applications. D) Elutions arent working well. May not be an issue—we often digest on the bead.
1 mg of lysate input is a good starting point to Co-IP from. You can expect about 20 ug of protein (target, binders, and background). Once your western blots reveal you pulled out the right protein, and a known interactor, you can try scaling down to include more conditions and replicates.
For nuclear POIs, we love the Dignam 1984 protocol. The salt extraction seems crazy, but it gives very clean results. We dont dialyze though, just slowyyy add the low salt buffer.
The same beads need to be used for the negative control IP (no or different affinity tag—recommended; protein KO, etc). Otherwise, the differential proteins IP’d by the different beads will give beautiful false positives.
On that note, you do not need a perfectly blank, protein-free background for a successful CoIP-MS experiment. In fact, we believe the background proteins prevent POI loss to tube adsorption, and provides a null distribution for statistical analyses.
Recommendations for proximity labeling, based on our experiences:
We turn to proximity labeling when either interacting proteins are low affinity and dont survive purification, or when you want to know what is close but not necessarily directly interacting. Think two TFs that sit next to each other on DNA but do not interact in solution.
The biotin ligase NOT fused to your POI needs to be subcellularlly localized the same as your fusion. Otherwise, you will get compartment specific enrichment which may look like statistically enriched proximal proteins.
When optimizing expression and labeling, its common for the biotin ligase alone to express higher and label more than the fused counterpart. This usually can be corrected for computationally, so dont worry too much.
Always check your fusion protein functions similarly, according to your biology.
Protein-protein interactions are commonly done to gain insight into the function of a poorly characterized proteins (guilt by association) or into mechanism. There are two major flavors of PPIs: proximity labeling and affinity purification/co-immunoprecipitation, both of which can be followed with quantitative mass spectrometry.
Recommendations for Co-IPs, based on our experiences:
We recommend starting in a standard lysis buffer like RIPA, that contains protease inhibitors, as well as any inhibitors for PTM hydrolases you may be interested in. You may need to try other buffers or lysis conditions but this is a great starting point.
In our hands, if you do not get a gDNA booger that makes the sample hard to pipette and doesnt spin down, the nuclei were not efficiently lysed, simply salt extracted. Try adding benzonase or, our fave, sonicating. This will lyse the nuclei and shear the gDNA.
Different affinity tags may need optimization. For example, the 0.1% SDS in normal RIPA can diminish anti-FLAG/DYDDDK IP efficiency. Monitor your flow-throughs, washes, elutions, and bead boils, to see where your protein of interest (POI) is.
Genomic Locus Proteomics
GLoPro is a technique to discover which proteins are associated with a specific genomic locus in living cells. It uses a catalytically dead CAS9 to localize the proximity labeler APEX2 to a predetermined locus of interest. This asks a very specific question and is different that general proximity labeling.
Space your guides 50-200 bp apart to make sure there is some overlap between labeling radii. We put use one guide per cell but some personal communications suggests several guides can be expressed in the same cell.
We havent explicitly tested how few guides one can use. But 4-5 feels like the right number. You will have to test 7-8 to find the best ones.
CAS9 binding seems to only preclude endogenous binding proteins within 10-20 bp of the protospace sequence. Keep that in mind if multiplexing guides.
The best quality controls are anti-FLAG ChIP to make sure your CASPEX is landing at the right site, and anti-biotin ChIP to make sure its labeling at the right site. The latter has proven challenging for a lot of labs. Unfortunately, there are not a lot of QCs after this before the big mass spec experiment, so we suggest being thorough. If you just cant get the anti-biotin ChIP to work, the next best is anti-FLAG ChIP confirmation, and low but inducible levels of biotinylation by western.
We also recommend that after you have made your engineered cell lines, to make sure your biology is not disrupted. It is not common but does happen.
We havent found the need to isolate nuclei….yet.


“lll finish the website soon, I promise”
— SAM